participants are not recruited based on specific diseases or traits, careful phenotyping is essential. Unfortunately, much of that clinical data was collected for billing purposes—not research—which can introduce noise and inaccuracies.
“We don’t want genetic discoveries to be skewed by data that was primarily collected for insurance billing,” said senior author Gamze Gürsoy, PhD, assistant professor of biomedical informatics. “We need to take the proper steps to make sure the data properly reflects the population before it can be meaningfully studied.”
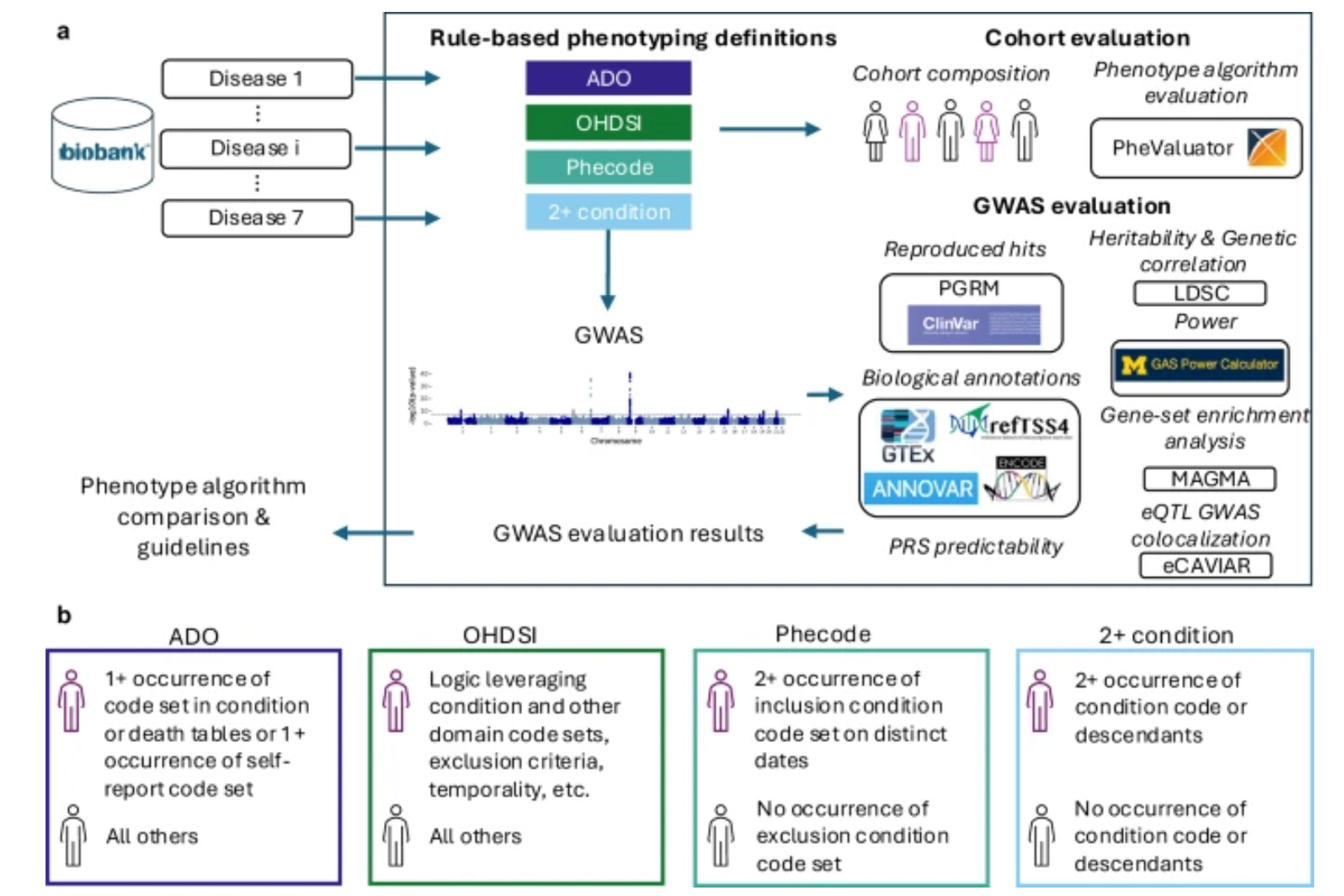
To address this challenge, Gürsoy’s team applied advanced rule-based phenotyping algorithms to improve the chances of correctly identifying who had the disease. The study, led by PhD student Abigail Newbury, was published in NPJ Digital Medicine and offers a roadmap for making real-world health data research more robust and biologically meaningful.
Defining Diseases
 “Phenotypes are the observable traits or conditions someone has—like asthma, diabetes, or even eye color,” said Gürsoy, senior author and assistant professor of biomedical informatics. “To find the genetic roots of diseases, we need to know exactly what we’re studying. If the labels are inaccurate, the genetic findings won’t be reliable.”
“Phenotypes are the observable traits or conditions someone has—like asthma, diabetes, or even eye color,” said Gürsoy, senior author and assistant professor of biomedical informatics. “To find the genetic roots of diseases, we need to know exactly what we’re studying. If the labels are inaccurate, the genetic findings won’t be reliable.”
Accurate phenotypes—identifying who does and doesn’t have a disease—are essential to reliable genetic discoveries. Mislabeled cases or controls can obscure genetic signals or mislead treatment strategies.
“Using overly simple definitions risks missing real cases or including people without the disease,” said Newbury, a PhD student in the Gürsoy Lab. “That weakens the genetic signals we’re trying to detect.”
To examine how phenotype definitions impacted genetic signal, the team compared three levels of algorithm complexity:
- Low complexity: relies on a single diagnosis code, and more specific codes that fall under it, recorded at least twice
- Medium complexity: includes more rules around diagnosis codes, including criteria to exclude unrelated conditions
- High complexity: relies on multiple data sources, including lab results, medications, diagnosis codes, and patient-reported disease, to create a more accurate picture
High Complexity, High Confidence
High-complexity algorithms outperformed the others: they identified more people as potential cases for seven tested diseases, found more genetic variants linked to those diseases, and produced results that reflected deeper connections to underlying genetic mechanisms.
“The high-complexity algorithms pulled from more parts of the patient record—not just diagnosis codes, but prescriptions, lab tests, and even self-reports,” Gürsoy said. “That gave a fuller picture of who actually had the disease, leading to stronger and more biologically meaningful genetic findings.”
Historically, researchers have often used low-complexity algorithms because they are easier to apply across databases. As Newbury notes, not only are high complexity ones more reliable, but they are becoming more practical as well.

“These simpler algorithms have been widely used in part because they are easier to construct and run on any database,” she said. “High-complexity algorithms traditionally had to be tailored to each database. Recently, large biobanks have started providing clinical data in a standard format, such as the OMOP Common Data Model. This makes it possible to run these more robust algorithms across studies.”
Making Treatment More Precise
High and medium complexity algorithms didn’t just identify more potential cases. They excluded individuals with similar or easily confused conditions.
For example:
- In type 2 diabetes, people with a prior diagnosis of type I diabetes or secondary diabetes were excluded.
- In asthma, people with a prior diagnosis of COPD or those taking medications commonly used to treat COPD were excluded.
“With more accurate case definitions, we can discover more meaningful genetic variants, build better risk scores, and eventually tailor treatment to the right subgroups of patients,” Gürsoy said. “It’s a foundational step for precision medicine.”
While high-complexity algorithms didn’t necessarily improve prediction models (who is likely to get a disease), they added significantly to the biological understanding of the condition, making future research and treatment strategies more targeted. As biobanks continue to expand globally, high-quality phenotype definitions will be critical for unlocking their full potential.
“Creating and sharing curated collections of high-quality phenotype algorithms, designed with clinician and researcher input and using data standards like the OMOP CDM, will help to drive research forward,” Newbury said. “These shared resources can help scientists around the world study disease more accurately and consistently, making it easier to uncover meaningful genetic links and, ultimately, improve patient care.”
Additional Details
The study, Multi-domain rule-based phenotyping algorithms enable improved GWAS signal, was published Aug. 2 in NPJ Digital Medicine.
All authors are from Columbia University: Abigail Newbury, Ahmed Elhussein, and Gamze Gürsoy.
This research has been conducted using the UK Biobank Resource under application number 100316. This work uses data provided by patients and collected by the NHS as part of their care and support. This study was funded by a Roy and Diana Vagelos Precision Medicine Award, a Warren Alpert Foundation award, and an NIH grant R35GM147004 to G.G. and an NIH grant T15LM007079 to A.N.